Laboratory of theoretical physical chemistry
3 | Quantum many-body molecular dynamics simulations aiming at a priori investigation of dynamical properties of quantum liquids and solids
It has been well known that light atoms and molecules such as hydrogen molecules, helium atoms, and protons in condensed matter at low temperatures obey not the principle of classical mechanics but that of quantum statistical mechanics because of their wave character. Condensed matter consisting of such quantum particles is called quantum liquids and solids whose properties are quite unique whereas they have not been so well revealed in the experiments as classical liquids. We have revealed dynamical properties of such quantum liquids and solids by means of the quantum many-body molecular dynamics simulations such as the path integral centroid molecular dynamics (PICMD), a quasi-classical approximation of the quantum statistical dynamics, with the emphasis on a priori prediction of the properties of the materials. A successful achievement of ours is the simulation-based prediction of the collective dynamics of liquid hydrogen (1990). The computationally predicted spectral profile of the dynamic structure factors of this quantum liquid have been proved a year later by a subsequent neutron inelastic scattering experiment performed in Europe. We have also succeeded in accurate evaluations of transport properties of liquid hydrogen by use of the PICMD simulations. The investigation of real-time dynamics of quantum condensed matter is still a cutting-edge subject, which we continue to pursue by means of quantum dynamics simulation methods.

Fig. 1 A snapshot of the spatial distribution of hydrogen molecules in liquid hydrogen at 14.7 K, calculated by the PICMD simulation. We can see that the distribution of each hydrogen molecule is spread like a bunch of grape because of the wave character of the molecule. Each ball in a bunch represents the coordinate of discretized imaginary-time path of that hydrogen molecule in the quantum regime. Each red ball represents the molecular centroid (or the center of the distribution) which evolves with real time by the quasi-classical framework of the PICMD.

Fig. 2 A snapshot of atomic spatial distribution of water molecules in ice Ic at 10 K, calculated by means of path integral molecular dynamics (PIMD) simulation. The spatial distribution of each atom is spread because of atomic wave character. Red balls: hydrogen atoms. Purple balls: oxygen atoms.
4 | Elucidation of reaction mechanism of molecular systems and development of new theoretical and computational method
We aim at elucidating essence of various chemical reaction dynamics involving electron, atom, and molecule using computational techniques based on quantum and classical mechanics. We are mainly focusing on two research themes; one treats the interaction of molecules with the laser field, and the other self-organization dynamics of the clusters consisting of carbon, boron, and nitrogen atoms. In the former, we are investigating the transition mechanism to a specified molecular state of a simple molecular solute in solvent molecules. We employ the mixed quantum-classical simulation method, in which we solve the time-dependent Schrodinger equation for a selected degree of freedom of the molecular solute and classically treat other degrees of freedom of the solute and solvent molecules. Especially, we are focusing on the adiabatic following processes using the STIRAP (Stimulated Raman adiabatic passage) and the chirped laser pulses. As for the latter, we are mainly investigating the growth mechanism of boron nitride fullerene using the simulation techniques that combines the quantum chemistry calculations such as the first-principle and semiempirical electronic state calculations with the action-derived molecular dynamics method.

Fig. 1 Vibrational excitation of hydrogen chloride molecule exposed to a tailored infrared laser pulse in the argon liquid. A mixed quantum-classical approach is used.
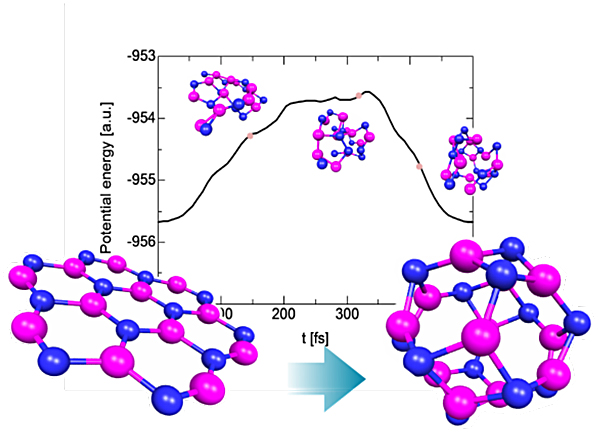
Fig. 2 Structural dynamics of B12N12 from a plane cluster to a caged cluster using the action derived quantum molecular dynamics method.



